-

Autoimmunity in membranous nephropathy
Clinical picture and causes of membranous nephropathy
Membranous nephropathy (MN, also membranous glomerulonephritis) is a chronic inflammatory disease of the kidney corpuscles (glomeruli) characterised by thickening of the glomerular capillary walls due to deposits of immune complexes (see illustration). These deposits damage the podocytes and impair the permeability of the glomerular basement membrane, leading to proteinuria. If protein excretion in the urine increases significantly (> 3.5 g / 24 h), nephrotic syndrome may develop, which is characterised by a reduced protein concentration in the blood (hypoproteinaemia), increased blood lipid levels (hyperlipidaemia) and oedema.1
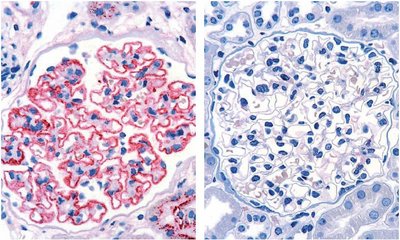 |
Approximately 20% to 30% of MN cases are secondary conditions (sMN), resulting from various underlying diseases or the use of certain medications.1, 2 These cases must be clearly distinguished from primary MN, which is caused by autoantibodies (see table). PLA2R and THSD7A are the first antigens to be discovered as target structures of these autoantibodies. If neither an underlying disease nor detectable autoantibodies are present, the condition is referred to as idiopathic MN.3
| Causes of pMN |
|---|
| Autoantibodies (PLA2R/THSD7A/NELL1) |
| Causes of sMN4 |
|---|
| Infections (e. g. hepatitis B, hepatitis C, syphilis, malaria) |
| Autoimmune diseases (e. g. rheumatoid arthritis, Sjögren’s syndrome, bullous pemphigoid) |
| Cancer |
| Other diseases (e. g. sickle cell anaemia, Guillain-Barré syndrome) |
| Drugs (e. g. nonsteroidal anti-inflammatory drugs (NSAID), gold, mercury, penicillamine) |
CONCLUSION: Since the treatment of the two disease forms differs significantly, the diagnostic differentiation between primary and secondary MN is of greatest clinical importance.4 In sMN, the therapy focuses on the underlying disease, whereas patients with pMN are mainly treated with immunosuppressives. Therefore, correct and fast diagnosis may prevent unnecessary diagnostic procedures or drug treatment.
Discovery and pathogenic value of the autoantibodies
In 2009, David J. Salant’s research group identified PLA2R, a 185 kDa glycoprotein, as a specific target antigen of autoantibodies that could be detected in 70 % to 80 % of all patients with MN without a secondary cause.3 In 2014, THSD7A was described as a second antigen associated with primary MN.5 The prevalence of antibodies against THSD7A is reported to be as high as 10 %. Although antibodies against both PLA2R and THSD7A can occur simultaneously in rare cases, anti-THSD7A antibodies are predominantly found in anti-PLA2R seronegative pMN patients.6 In 2020, NELL1, a third pMN-associated antigen, was discovered, which is considered to be the underlying autoantigen in up to 16 % of cases.9 In contrast, anti-PLA2R, anti-THSD7A and anti-NELL1 autoantibodies could not be detected in patients with sMN.
The antigens are expressed on the podocyte surface. They bind to circulating autoantibodies, forming immune complex deposits.
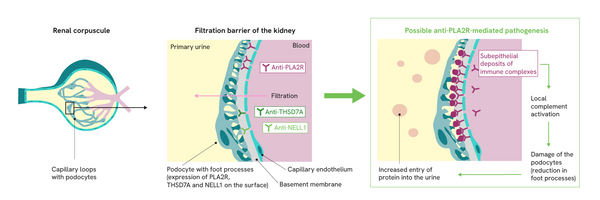 |
The pathogenic mechanism of the development of pMN is not yet fully understood. However, there is a consensus that the thickening of the glomerular capillary walls is caused by immune complex deposits. These deposits trigger local complement activations, which lead to a degradation of the cytoskeleton of the podocytes via the involvement of various mediators. As a result of this structural damage, the podocytes lose their filtration function and additionally secrete factors that further damage the glomerular basement membrane.1 This leads to progressive impairment of the filter function and thus to increased protein excretion in the primary urine (proteinuria) as well as the development of oedema and a compensatory increase in lipoprotein synthesis, which can result in nephrotic syndrome.
The relevance of autoantibodies in the pathogenesis of pMN was shown in 2017 for THSD7A in the mouse model: The injection of antibodies directed against THSD7A triggered pronounced nephrotic syndrome with proteinuria and hyperlipidaemia in mice.7
Although the pathomechanisms triggered by autoantibodies in anti-PLA2R- and anti-THSD7A-positive pMN are largely consistent, differences between the two patient groups have been observed in the clinical pictures. A study by Hoxha et al. (2016) showed a higher incidence of malignant tumours in anti-THSD7A-positive patients.8
1Ronco et al., Nat Rev Dis Primers 7, 69 (2021)
2KDIGO, Kidney inter. 100 (Suppl.): 1–276 (2021)
3Beck et al., N Engl J Med 361, 11-21 (2009)
4Trujillo et al., Nephron. 144, 261-271 (2020)
5Tomas et al., N Engl J Med 371, 2277-2287 (2014)
6Tomas et al., J Am Soc Nephrol 28, 3262-3277 (2017)
7Larsen et al., Mod Pathol 29, 421-426 (2016)
8Hoxha et al., J Am Soc Nephrol 28, 520-531 (2017)
9Sethi et al., Kidney inter. 97, 163-174 (2020)
Physicians and Laboratories
Information material
 Diagnosis without biopsy - Watch video now!
Diagnosis without biopsy - Watch video now!